
Lumpy作者对这个项目的定位与其说是做一个好用的工具, 不如说是构建一个通用的结构变异信号生成方案, 然后顺便对这个方案做了一个基本的实现. 与其他工具相比, lumpy的文章中花了更多的篇幅去阐述这个方案中设定的证据概念, 及获取信息的方案. 解读这个工具让我更加了解如何从NGS测序数据中获取结构变异的信号.
工具基本情况
lumpy使用C++编写, 所有的功能似乎都是自己实现的, 没有引用什么已有的模块. 软件可以从conda上获得, 程序本身近几年似乎没有更新, 所以不需要自己头疼从源码编译的问题.
软件检测结果格式可在VCF及BEDPE中进行选择. 不过需要注意的是, 两种格式包含的结果信息似乎有差异, VCF的会更详细一些. 另外本工具完全依赖BAM文件中对比对情况的记录, 因此如果比对本身的情况不理想, 必然影响下游结果. 然后, 工具本身不含有任何随机化的成分, 输入的文件一样, 检测结果就会是一模一样的.
Lumpy中的结构变异模型
Lumpy首先创建了一种结构变异断点模型: $b = (E,l,r,v)$
在这个模型中, 断点被定义为在待测基因组相邻但是在参考基因组上不相邻的一对基因坐标. 这一对断点通过四个指标进行描述, 分别是:
- $E$: 支持断点的一系列证据集合
- $l$: 左侧断点区间(interval), 包含区间的起止位点s和e(start/end)一个向量p, 这个向量p代表区间内每一个点是这个点是真正断点位置的相对概率(relative probability)
- $r$: 与$l$一致, 包含右侧断点的所有信息
- $v$: 断点复杂性(variety)信息, 主要包含不同证据的方向信息. 这个指标的存在是为了保留更多原始信息, 因为当结果被检出时, 只会标注其为某一类结构变异(如deletion), 但是原始的信号中可能包含支持其它类型的证据.
Lumpy对SR和RP信号都有使用, 每一个信号都能解析出一个断点的实例, 那么在读出所有断点实例后, 就需要根据包含的信息推断是否同一断点, 然后进行断点合并, 合并的条件是:
- 两个断点的两侧断点区间均有重合部分(阈值不详)
- 两个断点的复杂性一致
在满足上述条件后, 即可将两个断点合并起来, $E$中包含原先的两个信号, $v$不变(本来就一致), $l$和$r$则采取取算术均数(mean)的方式来合并, 即$l$的起点为原来两个起点的算术均数, 终点也是原来两个终点的算术均数, $r$也一样(但是多条时具体如何执行的未知, 两两依次去平均和直接取平均的结果是不一样的):
$$merge.start = \frac{l1.start + l2.start}{2}\ \ merge.end = \frac{l1.end + l2.end}{2}$$
根据作者描述, 这么做是为了减少离群证据对断点区间的影响. 证据合并完之后, 证据数目大于预设阈值的断点会被检测为结构变异. 确定检出的结构变异, 需要根据断点实例里包含的信息来确定, 以下是默认的计算方案:
- 断点的可能区间
- 区间内每个点是真实断点的可能性
首先, 变异的断点可能区间(interval)取断点实例的左侧断点的最大值, 和右侧断点的最小值(即取最小的可能区间).
而区间内每个点的可能性见下(以左侧断点为例):
$$l.p[i] = \prod_{e{\in}E}e.l.p[i-o]$$
- $i$是某一个基因坐标位置
- $l.p$是单个点是真断点的可能性向量, 前面的$l.$代表是左侧断点的
- $E$是所有证据的集合
- $e$是证据集合里面的每一个证据
- $e.l.p$单个证据里左侧断点的可能性向量
- $o$是偏移量(offset value), $o = e.l.s - s.l.s$, 即当前证据的左侧断点起点减去预检出的结构变异起点位置.
$e.l.p[i-o]$这一个略微有点绕, 其实本质跟变异位点的覆盖类似, 最终区间里每一个位置的p是证据里有这个位置的e的p值的积. 所以, 理论上最终区间中的某个点与越多的原始证据区间重合, 这个点的p值就越小, 对应就越可能是真的断点所在位置.
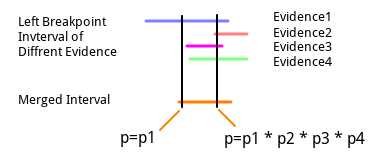
在进行证据合并的时候, 由于是不断的取算术均数来确定合并后的起止点的, 因此有可能最后的起点和止点中间是没有区间的(相等或颠倒?没有实例验证过), 在这种情况下, 作者会在所有的起点区间中, 找到坐标最大的那个坐标, 然后把没有经过这个坐标的区间删掉, 用剩下来的区间重新进行证据合并和区间以及概率向量的确定.(这一部分我不能完全确定, 因此原文贴出)
In the paper, Layer wrote:
Given the liberal merge process, it is possible for b.E to contain nonoverlapping distributions that would result in a zerolength product. In this case, we identify the maximum point among the sum of the distributions in b.E, any distribution not intersecting this point is removed, and the resulting subset processed normally.
如此一来, 就形成了最终的结果条目. 最终的结果条目中的断点区间还可以根据预设的百分比, 来进行缩小, 比如只取前99.9%等.
除了上述的方案, 还有一个Summation方案, 用于低质量数据, 这里暂时不解读.
从上面的方案可以看出, Lumpy本质上就是从bam里面读取含有信息的比对记录, 从记录抽取需要的信息, 然后根据信息解读结构变异. 那么一个关键的点就是从信号中抽取哪些信息. 为了定义需要的信息, Lumpy设计了一个叫做Breakpoint evidence的接口, 或者说通e用证据类:
这个接口要求下面3样信息:
- is_bp: 是否含有断点位置信息
- get_v: 结构变异的类型信息
- get_bpi: 断点的可能区间信息
也就是说, 对于每一个信号/证据, 都会解读出上述3样信息, 然后将这些信息做为一个单独的证据e, 加入到证据集E中, 进行之前说的解读过程.
那么如和从信号/证据中解读上述信息呢, 文章分别从SR, RP以及general breakpoint interface三类证据举例:
RP证据的解读
在双端测序中, Reads Pair内的插入片段是有一个正常的长度范围的, 且Read1和Read2的比对方向也是应该分别为+/-, 任何不正常的比对都提示了断点的存在, 因此解读出的信息如下:
- is_bp: 是, 可以确定断点存在
- get_v: 当两条Read在同一染色体上, 根据Read1和Read2的实际比对方向可推测变异类型(DEL/INV/DUP), 否则, 将统一为
inter-chromosomal - get_bpi: 通过两个Read的比对信息获取最终的断点区间以及区间的p, 有点复杂故见下
获得断点区间本质上就是根据比对信息从一条read的信息映射出断点的l(包含区间及区间中点的p值), 另一条read映射出r. 以l为例子:
- 根据Read的比对方向确定断点是在比对位置的上游还是下游
- 在进行判定前, 程序会通过从bam中抽取比对没有问题的Reads Pairs来估算插入片段的分布, 获取插入片段的平均长度$l$和标准偏差$s$(注意是标准偏差Standard Deviation, 非标准差), 另外要求一个人为指定的变量$v$, 以$l + vs$作为正常插入片段的预期数值. 由于打断时不太会出现超出预期的片段, 因此可以推测断点应该就在比对位置上游或下游$l + vs$(bp)长度的范围内, 也就有了断点的区间范围
- 对于上述区间内的每个点$i$, 其为真断点的可能性计算方式为$P(S(y).e-S(x).s > i - R(x).s)$(这里的$S()$原文没有明确解释, 我推断是sample的缩写, 与$R()$相对, 代表实际片段上的情况)
- $y$代表Read2, $x$对应Read1
- $S(y).e$是实际片段上Read2的终止位置, $S(x).s$就是实际片段上Read1的起始位置
- $S(y).e-S(x).s$即为实际片段的长度
- $i-R(x).s$即点$i$距离Read1比对起点的距离
- $P()$应当为从正常比对Reads获得的插入片段长度分布得到出的p值函数, $P(a>b)$的意义类似我们使用正态分布时里面的$P(Z>2.94)$, 使用对应分布的曲线下面积可以获得最后的p值, 从而评估这个位点是真正断点的可能性
SR证据解读
SR证据中, 一条read的相邻部分会比对到不相邻的基因组位置, 在lumpy中, 如果一条read存在多于两个小片段的比对, 则将这些小片段拆成两两小片段组分别考虑(比如三个片段就分$x_1,x_2$和$x_2,x_3$两组进行考虑), 从每一对小片段中解析出的信息如下:
- is_bp: 是, 可以确定断点存在
- get_v: 当两个片段的比对方向一致, 结构变异可能是DEL或者DUP中的一种(依据比对方向+/-不同), 比对方向不一致但在同一染色体上则为INV, 如果比对到不同的染色体则为inter-chromosomal
- get_bpi: 断点的区间位置与推测的结构变异类型有关. 考虑到测序中可能存在的错误, SR证据给出的断点位置也是有范围的, 这个范围大小取决于上述提到的输入参数$v$, 样品的质量(?)以及比对是使用的算法(?). 由于文献中对这部分的描述不如RP部分详细, 所以下面这些是个人的推论, 有空的话需要通过实际测试证实:
- 关于区间的获得: 理想的Split-Read断裂的两部分应该是相互不重叠的, 但是在实际中其实会存在两个部分有重叠的情况. 从lumpy作者自己写的SR Read提取脚本来看, 其确实是允许两个部分有一定长度的重叠的. 所以文中所说的SR证据的断点区间的与样品质量和比对算法有关可能就是指这个.
- 关于区间内的p值确定: 由于作者只说了这个区间的中点p值最小, 越靠近两边p值越大, 可能是内部分配了一个比例吧…这个可能需要自己造个证据bam一点一点去试了
general breakpoint interface解读
这种证据本质上就是记录了确定存在结构变异信息的BEDPE文件, 由于BEDPE文件中原本就会记录双边断点区间和结构变异的类型, 所以将这些信息与get_v, get_bpi对应即可. 关于BEDPE格式因为后面会有专门叙述所以这里略.
该类证据的存在作用我觉得有两个: 第一是在做遗传相关的分析时, 可以让人群中确定存在的变异更容易检出. 另一方面, 在做多工具同时检出时, 可将其他工具检出的比较可靠的变异结果与Lumpy的结果进行合并.