
“我所知道的结构变异相关信息”这个博客构想的内容有点太多了, 一次写太多我自己回查也不是很方便, 所以还是决定拆成各个小的部分, 之后再用gitbook写个综合的.
结构变异/拷贝数变异/基因融合
结构变异与SNP/InDel这两种小范围的基因突变相对, 泛指所有相对大规模的基因变异(具体多少bp以上算, 我暂时也不清楚). 大部分文献中这些变异都称为结构变异( Structural Variation), 这些变异被认为来源于基因组的重组(reorgnization)或称重排(rearrangement)这些变异的大小和样式比较多样了, 有生物课上讲过的整段染色体的交换, 也有单纯的片段缺失, 只是片段相对大而已.
目前对结构变异更多是按下面的类型来分:
- 插入/缺失(Insertion/Deletion, INS/DEL): 本质上就是InDel, 但是片段大
- 重复(Duplication, DUP): 代表某个区域连续重复出现, 另外重复下还有一种由’串联重复’(Tandem duplication), 待进一步考证
- 倒位(Inversion, INV): 即染色体内某区域的5’端和3’端颠倒过来
- 易位(Translocation, ITX, CTX): 即某个区段的转移, 可能是染色体内的转移, 也可能是染色体间的. 易位本身其实是一种复杂的结构变异. 比如染色体内易位, 某个片段从染色体上的A位置整段移动到了B, 那么A位置就会有这个片段的缺失, 而B位置则会有插入. 而如果片段插入的时候不是按原来方向插入的, 则同时也发生了倒位.
下面这幅图取自Gawroński的博士论文, 形象的展示了上述结构变异的类型.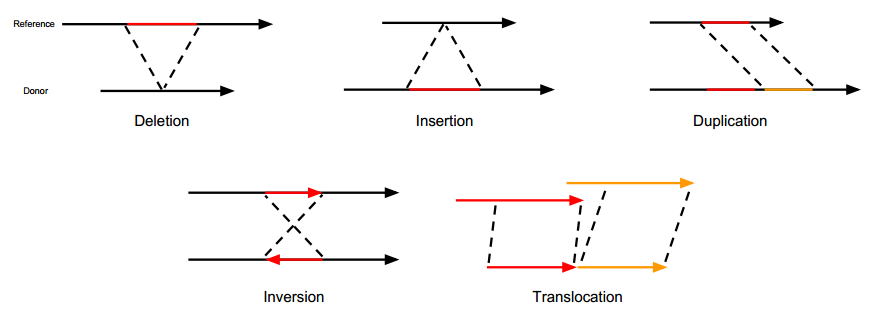
在结构变异的基础之上, 我们就可以进一步的来了解一下拷贝数变异(Copy Number Variation)以及基因融合(Gene Fusion).
拷贝数变异的关键是拷贝(Copy), 拷贝的概念呢…emmmmmm…这么说吧: 假设ATGCGGTAGCCGTAT是一个基因的完整序列, 我们就把这段序列称作一个拷贝. 然后人是双倍体, 所以正常来说, 一个人应该有两份这个基因, 也就是两个拷贝. 那么当基因检测的时候, 发现一个人的基因组里这个基因的拷贝数不为2, 我们就说检测到了这个基因的拷贝数变异.
由上面的概念可以看出, 拷贝数变异本质就是结构变异, 或者更确切一点, 是结构变异导致的一种结果. 如果基因组发生了缺失, 那么势必会有基因的拷贝数下降. 而重复则很明显会导致拷贝数增加. 那么, 为何拷贝数变异会被单独拿出来呢? 我个人认为, 这可能跟概念的提出时间或者其背后的实际意义有关. 假设每个不同拷贝的基因都具有一样的表达能力, 那么, 基因拷贝数的增加势必会使得基因的表达增加, 而拷贝数减少则会导致基因表达下降甚至相关表达产物消失. 简而言之, 与结构变异侧重于弄清楚基因区段到底发生了怎样的变化, 比如消失的区段从哪到哪开始, 消失的区段有没有去到基因组的某一个位置, 有的话这个位置在哪等具体的信息相比. 拷贝数变异关切的重点在于, 某个基因/区段的拷贝数是否变化了. 由于侧重点的不同, 拷贝数变异可以在结构变异检测中一并进行, 也可以单独进行, 当然实际上现在大多数时候是分开用不同策略进行的. 本篇主要还是侧重于结构变异, 所以后面的内容不会专门写拷贝数变异的相关内容.
与拷贝数变异类似, 基因融合(Gene Fusion)也是结构变异的结果. 比如某个基因区段整段缺失了, 那么区段的上下游必然会连接起来. 如果消失区段的前后两个断点刚好都在两个基因的表达区域内, 那么这两个基因就被连接在了一起, 这就是基因融合. 所以我们可以看出, 所有的结构变异都有可能导致基因的融合, 只是最终融合起来的方式不太一样. 基因融合受到关注由一些疾病/癌症的病因确认有关. 比如癌症中的EML4-ALK融合, 慢性粒细胞白血病中的BCR-ABL融合. 由于基因融合关注的是最后是否有基因连在了一起, 以及怎么连接的(这关乎基因表达是上升还是下降了), 所以其在检测的时候也有专门的策略与工具, 详细见后.
使用NGS数据检测结构变异/基因融合
二代基因组重测序的基本原理是把完整的基因序列打碎, 对碎片序列进行测序后将其比对回参考基因组后重组序列的原貌. 在这个比对的过程中会产生一些”不正常”的比对结果, 这些结果中, 有些能提示我们待测基因组相对参考基因组发生了结构变异, 我们的检测也就是基于这些比对结果, 我们将之称为结构变异信号(signal)/证据(evidence)
结构变异检测策略
结构变异检测的基本策略一般归类为4类, 一类一类来:
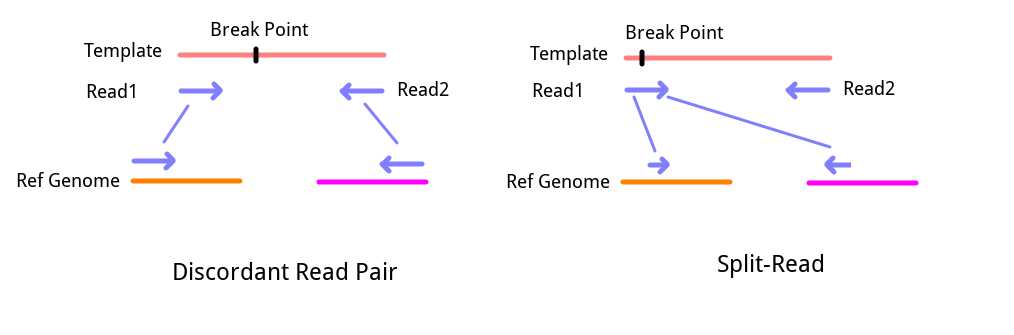
1. Split-Reads
如果之前有接触过NGS的序列比对文件(SAM/BAM), 一定在文件中见过Clip-Reads. 简单来说, 这些序列在比对的时候并不能全长比对到基因组的某个地方, 而是一部分比对到一个地方, 另一部分比对到另外一个地方. 这些序列可能来自于刚好包含了结构变异断点的序列, 因此才会出现这种异常的”嵌合比对(Chimeric alignment)“. 通过将这些序列抽取出来, 并对两处Clip序列的比对情况进行回溯, 可以一定程度长推测发生的结构变异类型. 而基于这种信息进行结构变异检测的策略就是SR策略.
2. Discordant Read Pairs
如果采用了双端测序(Pair-End Sequencing), 那么测序结果将是成对出现的. 根据测序原理可知, 双端测序得到的是一个DNA单链5’端和3’端的序列. 由于测序文库制备时, 会将DNA打断到一个固定的范围内, 因此, 正常情况下一对序列的Read1和Read2在基因组上的距离应该是出于一个固定范围内, 并且应该是一条正向比对上参考基因组, 另一条则是反向互补比对. 但是在实际的比对结果中, 总会有一些序列对不符合上述情况, 比如比对距离相去甚远甚至不在同个染色体上, 或者Read1 Read2都正向比对到基因组上等等. 在这种情况下, 这一对序列就称作”Discordant Read Pairs”, 可能由于这种信号来自一对序列, 所以Lumpy中直接称其为”Pair-End Evidence, PE”, 但基于这种信号的检测策略好像更经常被RP(Read Pair)策略(这个需要之后多看一点评测文章确认). 这样的Reads对同样可能来自包含了结构变异断点的序列, 区别则在于断点的位置. 对于Split-Read, 断点位于序列一端, 所以刚好被测出来了. 而对于Discordant Read Pairs, 断点应该在插入片段范围内, 也就是并没有直接体现在测得的序列上.
3. Read Count或者叫Read Depth
这种策略就如其名字, 是基于特定片段的测序深度的检测方案. 很明显, 这是用来检测拷贝数变异的. 不论发生了何种结构变异, 如果最终导致了某基因的拷贝数减少, 那应该会直观反应到该基因的比对结果上—-该基因区域的测序深度明显下降. 反之, 如果发生了拷贝数增加, 测序深度就应该相应增加才是. 这个策略从原理上说来相当简单, 但是在实际应用的时候并不会必前面的两个策略简单多少. 因为其面临一个说来简单但非常麻烦的问题: 深度高多少算明显高, 低多少算明显少? 这个问题使用外显子或目标区域测序进行拷贝数变异检测时会变得更为复杂.
4. de novo Assembly
这里的de novo Assembly和二代测序中的不依赖参考序列, 从头组装基因组其实是一个意思. 应用在结构变异检测上, 则是在查看SR或者PE信号之前, 先对获取下来的序列进行组装. 因为序列组装后获得更长的片段, 这些使用这些片段再次进行比对会获得比短序列更好的结果. 可以看出这种简称AS的策略不会单独使用, 因为其目的在于获取更长的片段, 重新比对后再按照SR的思路来进行检测.
关于检测策略的个人思考
1. 测序方案的理论和现实
就象黄树嘉老师博文中写的那样, 想获得更可靠的结构变异检测结果, 提高片段长度, 进行全基因组测序是最简单直接的方式. 然而这两种方案都意味着单次检测成本的增加, 在测序的成本真正白菜价以前, 使用尽可能少的数据来获取更可靠的结果必然是结构变异检测应用在临检领域所必须面临的问题.
2. PE和SR信号的获取难度的差异
假设一个结构变异断点周围的序列被测出来的可能性是相等的, 那么出现PE信号的可能应该是大于SR信号的, 因为测序时插入片段的长度往往是比Read本身要长, 自然断点位于插入片段区域的时候会更多一些. 另外, 由于大多数检测手段还是要基于比对, 那么一些SR信号还可能会因为断点位置不好(太靠近Read两端), 难以正确比对而导致信号丢失. 所以, 在同样的测序深度下, 尽量去应用PE信号理论上是能提高检测的灵敏度的.
3. 基于比对带来的检测瓶颈
由于大部分工具都还是要基于序列的比对情况的, 因此如果真实的变异信号存在比对困难(序列太短无法比对), 必然会导致一些信号的丢失而影响检出. 这也催生了一些不基于比对的检测方案的尝试.
基因融合专属检测策略
在前面我们已经说过, 基因融合是结构变异的一种结果. 某种基因融合受关注, 多半是因为这种融合会导致基因表达的异常, 进而产生疾病. 因此在基因融合的检测中, 侧重点在于检测是否有基因融合在一起了, 以及它是怎么融合的. 由于这个侧重点的不同, 除了使用结构变异的检测策略从基因组上检出结构变异然后再进行融合可能的解读外, 也衍生出了专门进行融合检测的方案: RNA-Seq融合检测. 这种方案直接在转录组中进行融合检测, 如果检测到了来自与不同基因转录本的序列, 即可以说明存在基因的融合事件. 同时, 由于检测的是RNA, 也不用费力去推测融合会不会发生, 因为检测到即证明了融合的有效性.
当然, 个人认为这种方式也不是十全十美的, 能想到的缺点有如下:
- 检测难度较DNA高, 毕竟RNA易降解…
- 作为应用于临床检测的方案, 使用RNA检测应该会有样品的限制和成本上的限制
由于我暂时没有在实际工作中使用过这类工具, 也暂时没有使用的计划, 所以这个部分等下文的坑填完了再回来挖坑.
另外, 目前大多数结构变异/融合的检出方案中, 向参考基因组比对都是必不可少的, 然而这个过程目前来说比较耗时的(使用了FGPA芯片做专门优化除外). 而基因融合本质上只需要了解是否有序列显示两个基因融合在一起了, 因此, 目前也见到有商业公司开发了基于非比对数据(fastq)的检测方案(INVALID POST SLUG PROVIDED ), 该方案但从检测速度来说确实快了非常多.
最后还有一点要注意, 由于侧重点的不同, 一些专门的基因融合检测软件在汇报融合结果的时候只汇报融合的基因及具体的外显子, 其形式多种多样, 无法直接与结构变异的检测工具进行比较.
工具
早期的结构变异软件会使用前述的一到两个检测策略, 而比较新的检测软件基本都是多策略的了…物尽其用嘛. 我之后解读的几个工具都是相对较新的, 他们在策略上都是多策略并用的. 其中Creast是我第一个使用较多的结构变异检测工具, 对结构变异的初步了解也是来自它, 不过它的年份也是相对久远了… Lumpy是我第二份工作中首要接触的工具. SvABA和SViCT是两个我在Github上找到的工具, 都比较新, 前者是2018年发布的, 是一个与Broad Insititute合作的项目, 后者2019年发布, 号称是第一个针对cfDNA设计的结构变异检测工具. 这里目前挖坑的都是我有实际用过的工具. 对于我在工作中见人提起的比较多的Manta, Delly以及后续新看到的工具, 如果有必要的话我再慢慢补充.