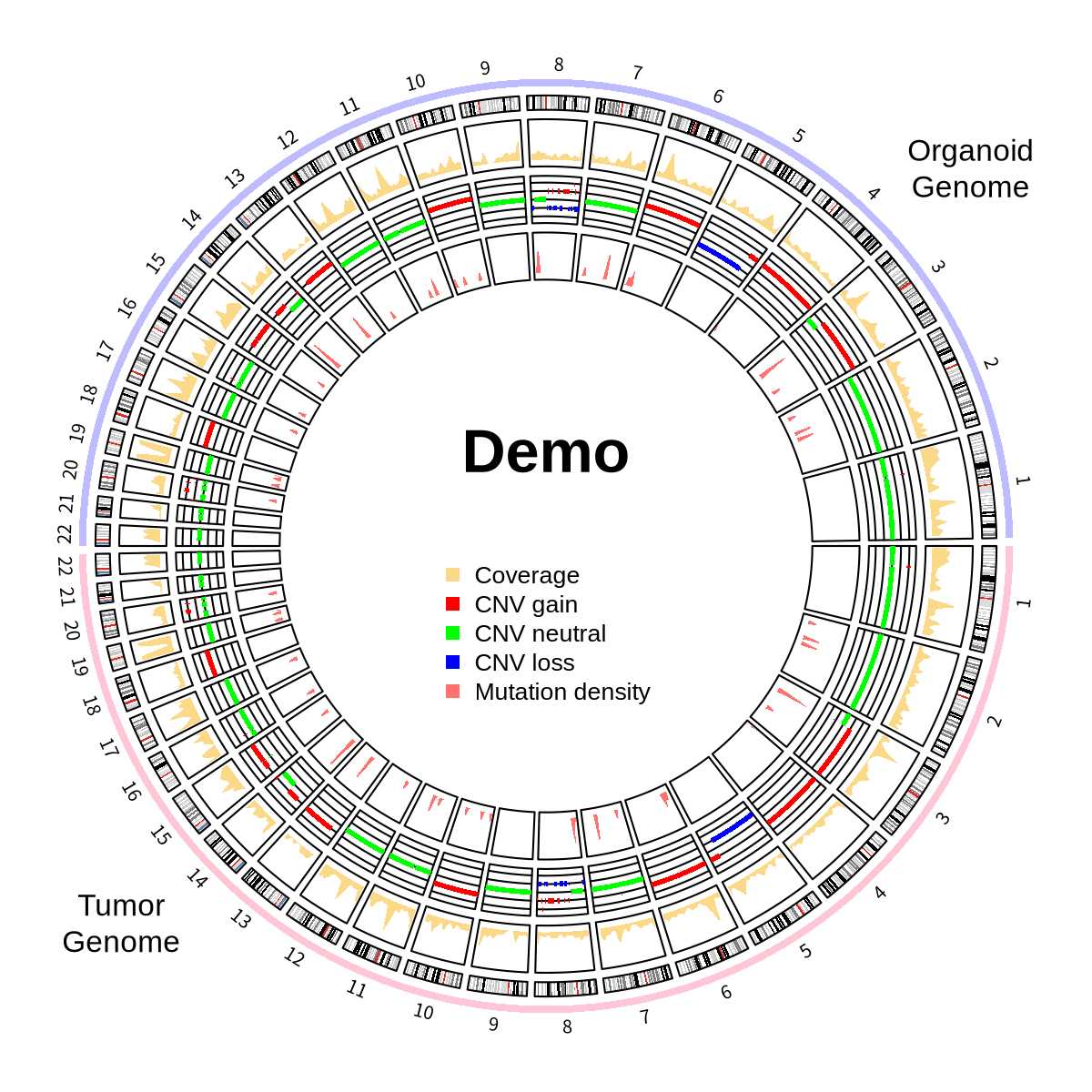
In cancer research, comparing genomic features between tumor samples and organoid models is crucial for validating model reliability. Circos plots provide an intuitive way to visualize detected mutations across the genome, making them commonly used for representing overall detection results of representative samples. While reading the circlize documentation, I came across an example demonstrating paired samples, which I found suitable for showcasing paired primary samples and organoids. I’ve adapted it to create a plot for displaying paired samples. The code is primarily based on the official documentation’s 9.5 Concatenating two genomes
Preparation
Loading Necessary R Packages
First, we need to load some essential R packages:
1 | library(circlize) |
Among these, MutationalPatterns is mainly used for loading VCF files to avoid manual processing, while BSgenome.Hsapiens.UCSC.hg38 provides the necessary information for drawing basic chromosomal regions.
Data Preparation
This visualization showcases three types of information:
- Somatic mutation detection results (VCF files)
- CNV detection results (from CNVKit)
- Sequencing coverage data (directly using results from CNV software to avoid duplication)
Building Genomic Framework Data
We first construct a combined framework containing both tumor and organoid genomes:
1 | # Remove XY chromosomes and build combined genomic data |
This creates a data frame named cytoband that contains basic information about the two genomes to be visualized.
Processing Mutation Data
Using the MutationalPatterns package to read VCF files and convert them to a suitable format for plotting:
1 | vcfs <- read_vcfs_as_granges( |
Processing CNV and Coverage Data
Both CNV and coverage data are obtained from CNVkit:
1 | # CNV data |
Drawing the Circos Plot
Setting Colors and Initialization
1 | # Color settings |
Adding Chromosome Tracks
1 | # Set chromosome track |
Adding Coverage, CNV, and Mutation Density Tracks
1 | # Sequencing coverage |
Adding Text and Legend
1 | # Set center text |
Complete Code
1 | library(circlize) |
Final Result
This uses data generated from one sample to create both the upper and lower circles, so the detection results appear identical on both sides. In reality, if paired results were this consistent, it would be quite remarkable.